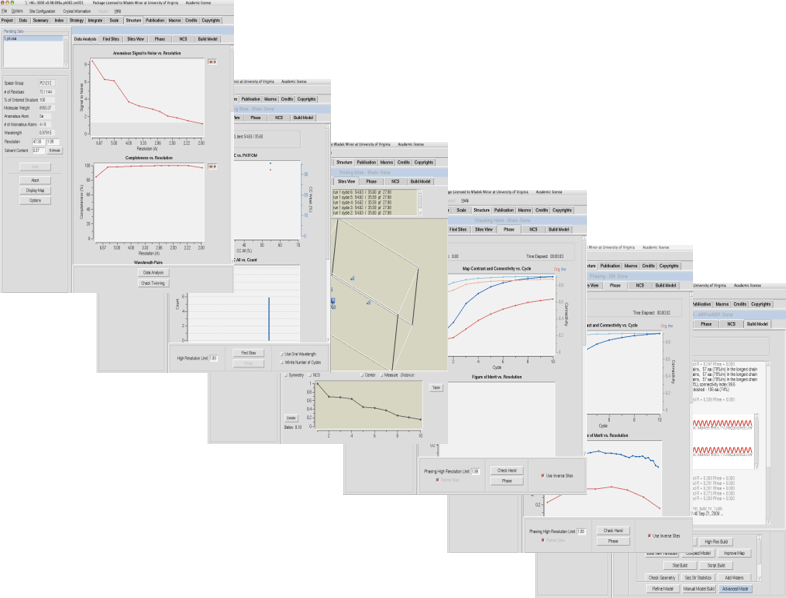
HKL-3000
Available from HKL Research, Inc.The HKL-3000 software suite provides a seamless path from raw X-ray diffraction images to a refined structural model. It combines the data collection and reduction functions of HKL-2000 with tools for data analysis, substructure solution, phasing, and model building for macromolecular crystallography. It merges different crystallographic software into a complete structure solution pipeline for both SAD/MAD and molecular replacement (MR) phasing.
HKL-3000 is built on years of experience solving many structures (including both straightforward and difficult cases) by macromolecular crystallography. Based on that experience, the default algorithms and parameters for each step of the process were chosen to be optimal for the majority of structure solution problems. For most straightforward cases, you should find that the default settings will not need to be changed to solve your structure. However, many procedures of HKL-3000 have an “Advanced Mode” dialog that permits you to change the default parameters (and in some cases choose the algorithm/programs used) as needed to solve more difficult structures.
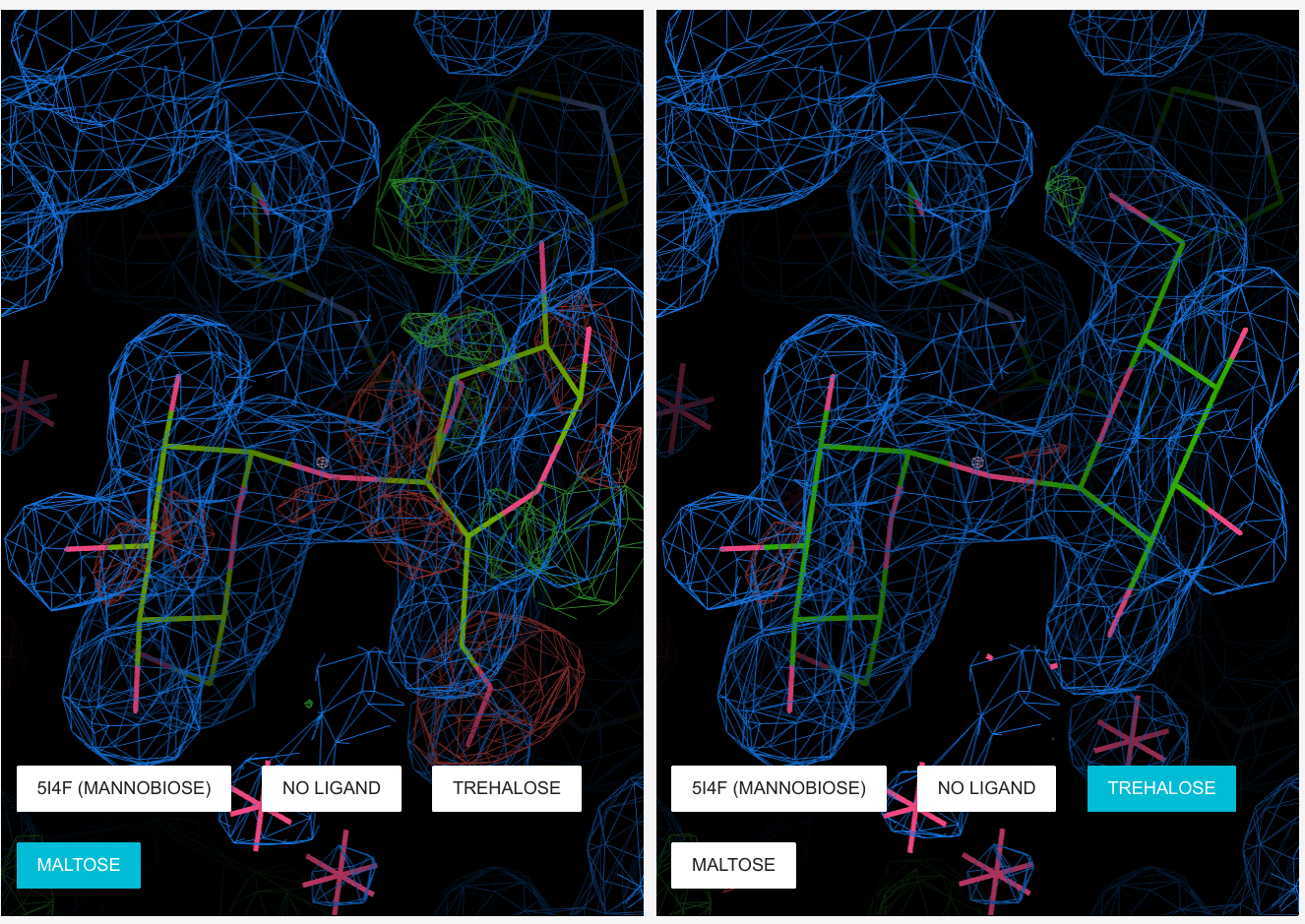
MolStack
Molstack ServerMolstack is a platform for structural biologists that allows the presentation of pre-selected views of a structure and corresponding electron density. Components of each view can be turned on and off and two different depictions of a view can be displayed side by side, allowing a direct comparison of different models and/or maps. This allows structural biologists to present their interpretations of a structure along with the experimental evidence upon which they based their conclusions.
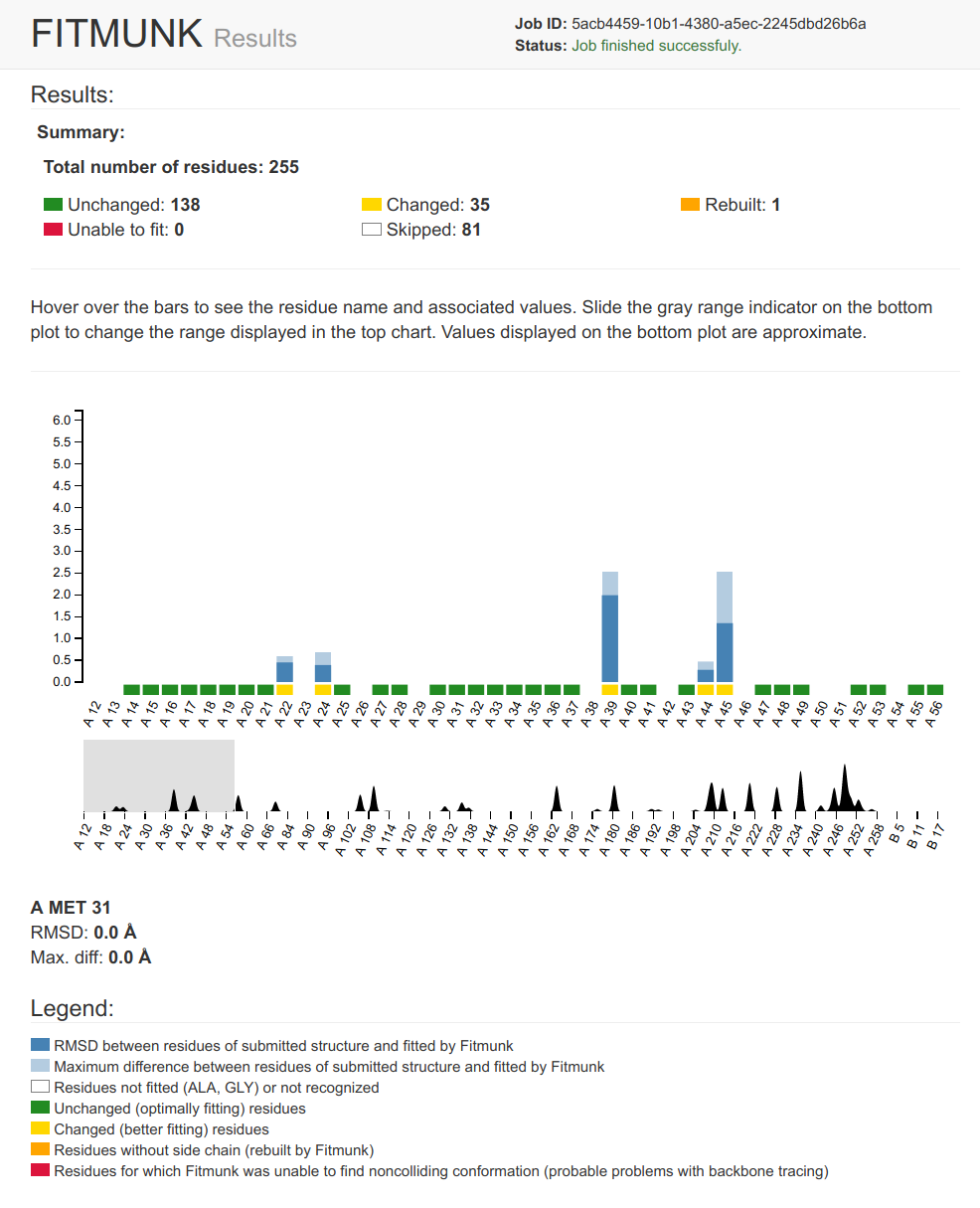
Fitmunk
Fitmunk ServerFitMunk is a online tool designed to automatically improve the accuracy of side chain placement. Using extensive conformational sampling and a novel hybrid energy function, Fitmunk can help improve the quality of crystallographic models, especially at medium and low resolutions. Although particularly useful during the early stages of refinement, Fitmunk can also be used to validate existing structures.
ProteinDiffraction.org
ProteinDiffration.org (IRRMC)Officially known as the Integrated Resource for Reproducibility in Macromolecular Crystallography (IRRMC), proteindiffraction.org provides a means for crystallographers to archive their raw diffraction images. The site currently maintains over 6400 datasets for over 3400 projects as well as the metadata to allow processing the data. This resource also allows archival of unsolved data sets, which can permit data solution when molecular replacement models become available or newer crystallographic algorithms can solve the phase problem.

CheckMyMetal
CheckMyMetal ServerCheckMyMetal (CMM) is a validation tool for metals bound to macromolecules. The validation parameters that CMM examines cover the entire binding environment of the metal ion, including the position, charge and type of atoms and residues surrounding the metal. CMM can detect discrepancies from target values of the parameters that it assesses, and highlight potential problems in metal assignment and modeling. Hence, CMM is a convenient validation tool that is complementary to existing experimental methods for metal identification in macromolecular structures. CheckMyMetal can evaluate user submitted structures and has pre-calculated results for structures in the PDB.
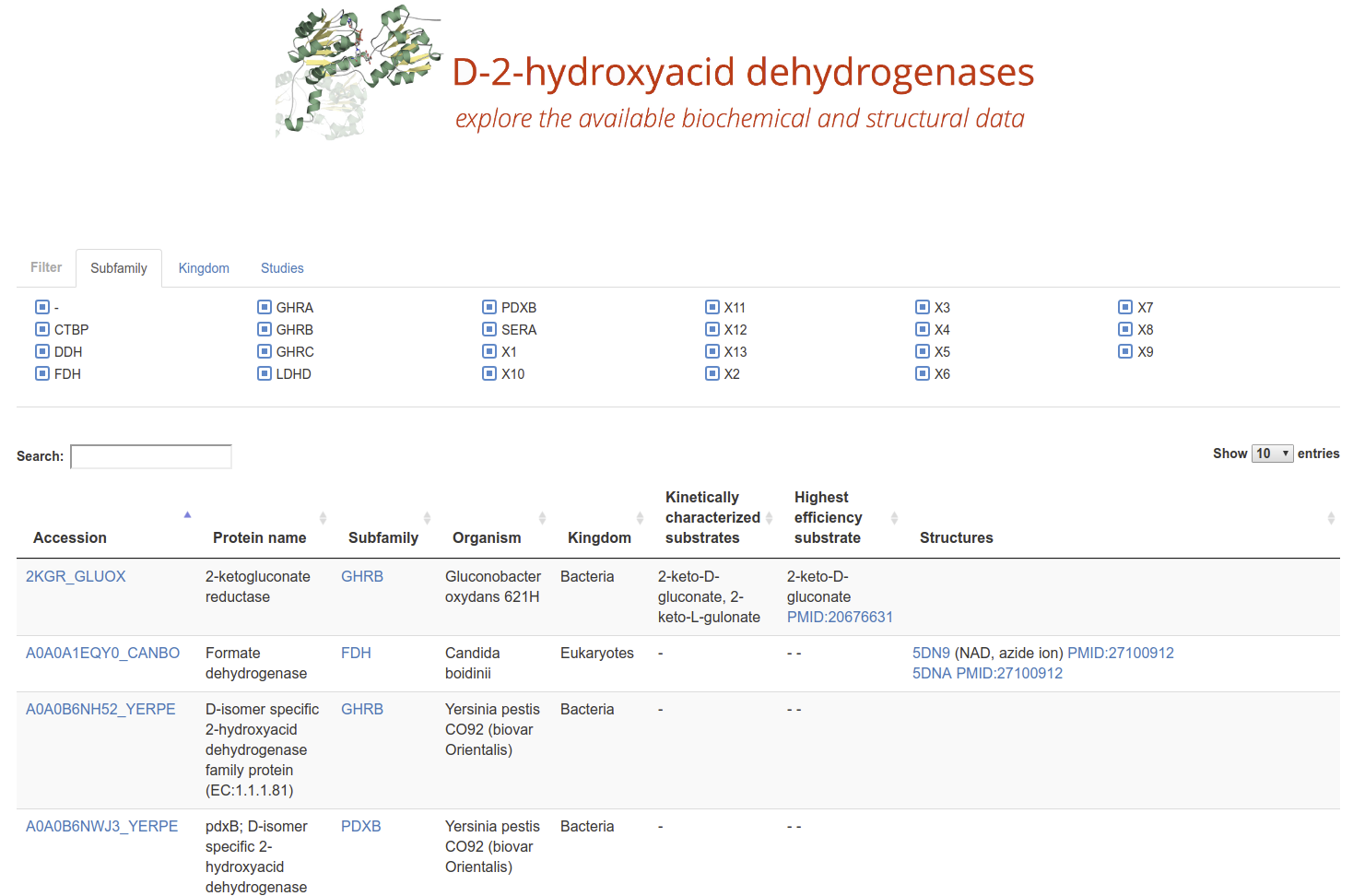
D-2-Hydroxyacid Dehydrogenases
2HADH Knowledgebase The D-isomer specific 2-hydroxyacid dehydrogenases (2HADHs) comprise a diverse family of oxidoreductases with various metabolic roles and biotechnological applications. The complex evolution and broad sequence diversity of this family hinder functional annotations for uncharacterized members. To simplify 2HADHs classification, we have created a 2HADH knowledgebase that consists of three elements: an explorable phylogenetic tree of the family, an interactive table with annotations of the selected enzymes, and a BLAST search tool.
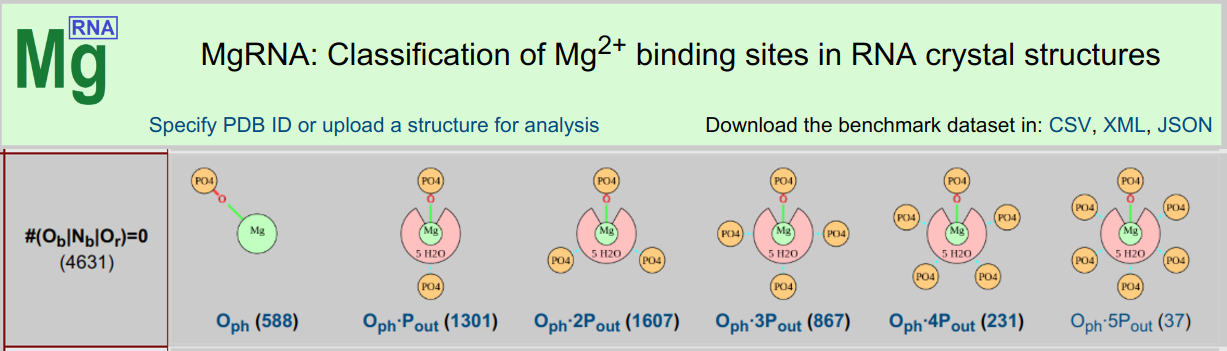
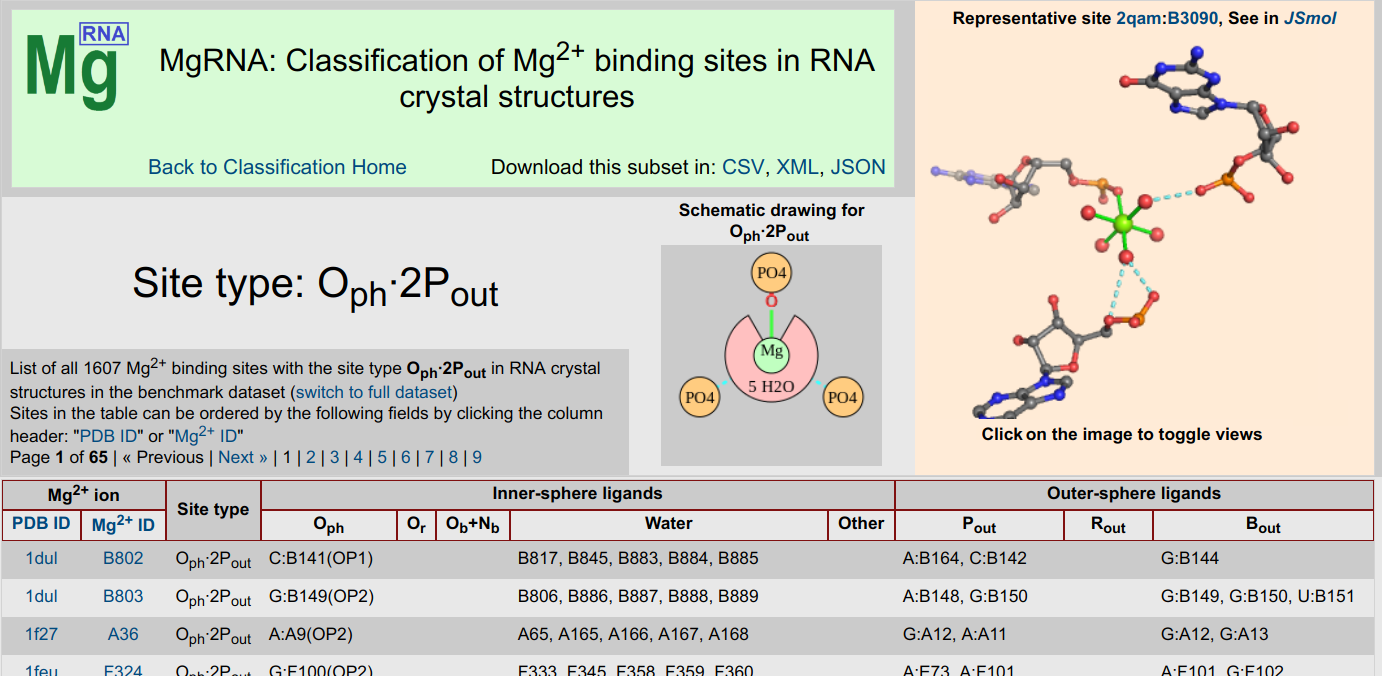
MgRNA
MgRNA ServerMgRNA provides a classification scheme for Mg2+ binding sites in RNA. After a thorough analysis of the Mg2+/RNA structures in the PDB, discrete motifs of Mg2+ binding were identified. The server can be used to analyze user-submitted structures and has pre-calculated results for structures in the PDB.
LabDB
LabDBLabDB is the Laboratory Information Management System (LIMS) designed by the Minor Lab to keep track of the experimental details associated with the purification, crystallization, and structure determination of proteins.

